
Trois étapes pour la traçabilité de la qualité et de la conformité du développement de logiciels de dispositifs médicaux

De nombreux dispositifs médicaux contiennent des logiciels complexes et des interdépendances avec d’autres systèmes logiciels et matériels qui n’ont pas été conçus pour fonctionner dans un environnement critique pour la sécurité. [Illustration via Adobe Stock]
Un ancien évaluateur de la FDA offre son point de vue sur la manière de renforcer la confiance dans la validation des logiciels médicaux.
Par Paul Jones, Ketryx
Lors de l’examen des logiciels dans les logiciels en tant que dispositifs médicaux (SaMD), les logiciels dans les dispositifs médicaux (SiMD) et les systèmes de systèmes de dispositifs médicaux (SosMD) à la FDA, j’étais toujours à la recherche de preuves suffisantes pour justifier l’affirmation des sponsors selon laquelle l’appareil fonctionne comme prévu, de manière sûre et efficace.
Les arguments à l’appui de cette affirmation découlent des artefacts du processus d’assurance qualité, de contrôle de conception et d’action corrective et préventive (CAPA) du système de gestion de la qualité (QMS) du fabricant, qui incluent la distribution des appareils, la surveillance après commercialisation et les mises à jour.
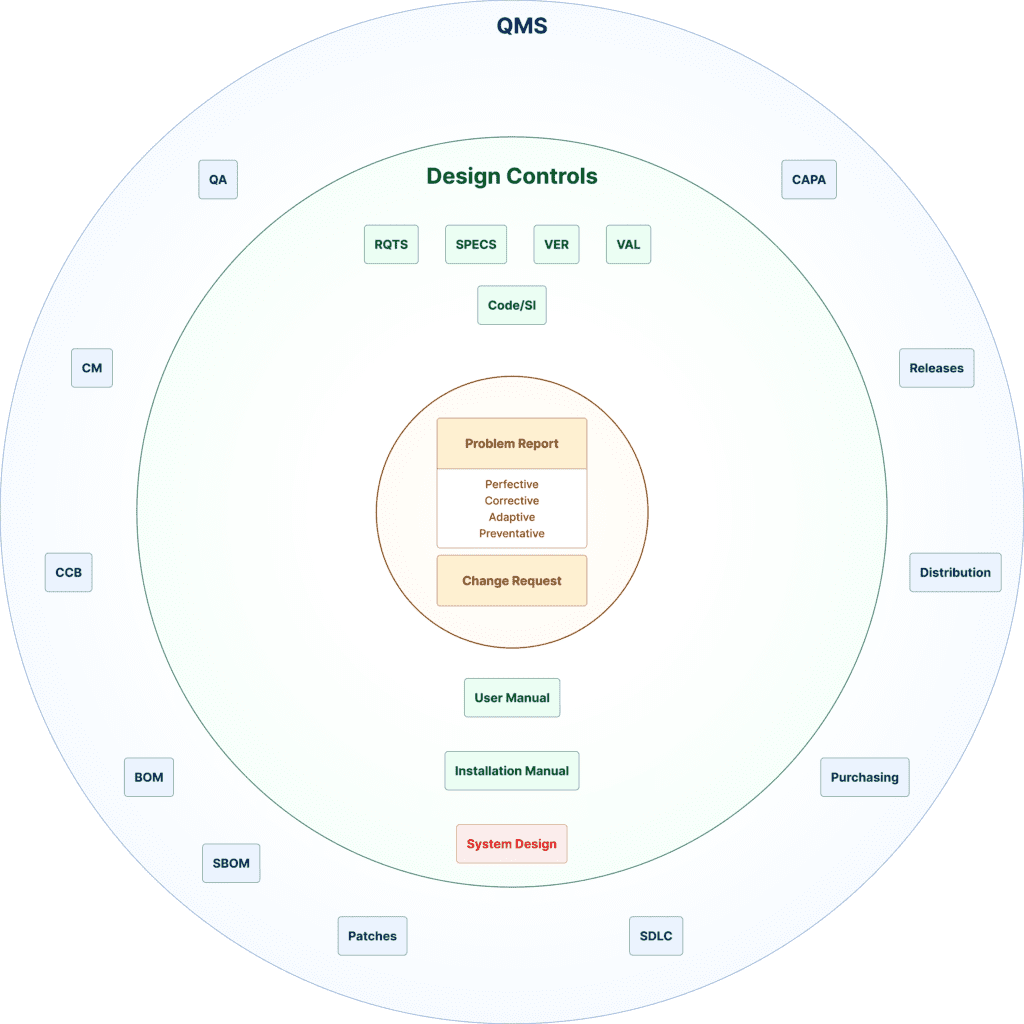
[Courtesy of Ketryx]
Le concept de traçabilité entre ces artefacts était essentiel pour établir ma confiance dans les allégations de sécurité et d’efficacité. Pour comprendre pourquoi, vous devez d’abord comprendre comment une application logicielle (ou la plupart des produits) est créée.
Cela commence par les exigences relatives à l’appareil : ce que l’appareil est censé faire dans le contexte de l’utilisation prévue, comment l’appareil interagira avec ses utilisateurs et ses patients, quel est l’environnement, ainsi que les risques et les mesures d’atténuation en matière de sûreté et de sécurité. À partir de ces exigences, spécifications, logiciels les activités de développement et de test sont effectuées, aboutissant à un système logiciel validé.
Si vous ne pouvez pas établir de traces entre les différents éléments de ces activités, vous ne pouvez pas établir un argument crédible selon lequel l’appareil fonctionnera comme prévu. Par exemple, s’il n’existe aucune spécification de conception correspondant à une exigence de conception, il est probable que cette exigence ne soit pas incluse dans le produit final et que le dispositif ne fonctionnera pas comme prévu.
Lorsque j’examinais les logiciels des appareils, je constatais souvent de mauvaises propriétés d’exigences/spécifications qui se répartissaient en plusieurs catégories. Ils étaient ambigus, non testables, peu clairs ou peu concis. Les documents de contrôle de conception n’avaient pas d’identifiants uniques pour prendre en charge la traçabilité, manquaient de signatures, répertoriaient des tests incomplets, manquaient de mesures de qualité logicielle ou de statut de défaut/anomalie, et avaient des tableaux de traçabilité incomplets ou inutiles.
Trop souvent, les fabricants n’ont pas utilisé plusieurs méthodes complémentaires d’analyse des risques dans leur processus de gestion des risques. Lorsque j’examinais des artefacts présentant ce type de déficits de qualité, il était difficile d’avoir une grande confiance dans le comportement de l’appareil, et je demandais souvent des informations supplémentaires (lettre AI) qui augmentaient le délai de mise sur le marché du fabricant.
La combinaison de contrôles d’assurance qualité médiocres et de contrôles de conception médiocres a érodé ma confiance dans la capacité du fabricant à assurer de manière adéquate la qualité des appareils tout au long de leur durée de vie (contrôle total du cycle de vie du produit, ou TPLC). TPLC englobe le développement de produits, les chaînes d’approvisionnement, la distribution, la maintenance et les activités de fin de vie. Si la confiance des évaluateurs est faible, ils peuvent rejeter la soumission.
1. Comprendre les directives de la FDA
Le 14 juin 2023, la FDA a remplacé la version de mai 2005 du Guide relatif au contenu des soumissions préalables à la commercialisation des logiciels contenus dans les dispositifs médicaux par le Contenu de la soumission préalable à la commercialisation pour les fonctions logicielles des appareils. Il existe des différences majeures dans les informations que la FDA s’attend à soumettre entre ces deux documents.
Le changement le plus important concerne le passage d’un niveau de documentation à trois niveaux à un niveau de documentation à deux niveaux, basé sur le risque dans le contexte de l’utilisation prévue de l’appareil. Une multitude de documents d’orientation relatifs aux logiciels alimentent les nouvelles lignes directrices sur les fonctions logicielles qui traitent des produits d’appareils à fonctions multiples, de l’utilisation de logiciels disponibles dans le commerce, de la validation des logiciels, des facteurs humains, de la cybersécurité et d’autres fonctions et facteurs. En termes simples, le promoteur est censé soumettre les informations spécifiées dans toutes les directives pertinentes.
Les nouvelles lignes directrices autorisent une déclaration de conformité à la norme ISO 62304 pour les pratiques de gestion de configuration et de maintenance au lieu de certaines documents spécifiés.
2. Anticiper les attentes des évaluateurs
L’examinateur de la FDA s’attend à voir la documentation spécifiée dans toutes les directives relatives au logiciel, en utilisant une liste de contrôle pour vérifier que la documentation soumise est complète. S’il manque quelque chose, l’examen peut être retardé jusqu’à ce que la documentation soit fournie.
En général, l’examinateur du logiciel examinera les documents soumis pour prouver qu’il existe un bon système qualité, que les risques connus et prévisibles sont pris en compte et que le logiciel est validé conformément aux exigences des normes ISO 62304 et ISO 14971.
Les orientations liées à la cybersécurité ajoutent considérablement plus d’attentes en matière de documentation de la part du réviseur. Les examinateurs de la FDA accordent de plus en plus d’attention aux problèmes de cybersécurité avant et après la commercialisation. Les fabricants d’appareils s’appuient de plus en plus sur Internet, le cloud computing et les réseaux pour fournir des services innovants aux patients. La menace d’événements de cybersécurité a conduit à une prise de conscience accrue de l’importance de disposer d’un TPLC de qualité.
Des vulnérabilités peuvent être introduites n’importe où dans le processus TPLC. Il est crucial de pouvoir maintenir la traçabilité au sein de la chaîne d’approvisionnement, du développement des produits, de la production, de la distribution et des mises à jour. Les incohérences peuvent nuire aux patients et aux écosystèmes de santé.
3. Préparer votre soumission
De nombreux dispositifs médicaux contiennent des logiciels complexes et des interdépendances avec d’autres systèmes logiciels et matériels qui n’ont pas été conçus pour fonctionner dans un environnement critique pour la sécurité. Cette complexité rend difficile, voire impossible, le recours à des activités manuelles telles que le suivi des progrès dans des feuilles de calcul. La nature complexe du développement logiciel nécessite un système de gestion de la qualité sophistiqué capable de gérer cette complexité.
Les tâches telles que le contrôle de la documentation, la gestion des changements, les activités de vérification et de validation ont dépassé les limites des approches manuelles traditionnelles. La solution réside dans l’exploitation de la puissance de calcul pour gérer le processus TPLC. En tirant parti des capacités informatiques, les informations du processus TPLC peuvent être systématiquement organisées, authentifiées, vérifiées et préparées pour les soumissions réglementaires et la livraison aux clients avec un niveau de confiance élevé.

Paul Jones est vice-président des affaires réglementaires/assurance qualité chez Ketryx. [Photo courtesy of Ketryx]
Paul Jones, vice-président des affaires réglementaires/assurance qualité chez Ketryx, a contribué à créer l’approche de la FDA en matière de logiciels et de dispositifs médicaux critiques pour la sécurité et a fondé le laboratoire d’ingénierie logicielle de la FDA. Pendant près de 25 ans au sein de l’agence, il a examiné plus de 300 appareils, occupé des postes au sein de comités au sein de groupes qui traitaient des normes de sécurité des logiciels médicaux comme ISO 13485, ISO/IEC 62304 et ISO 14971, effectué des inspections et formé le personnel de la FDA sur la qualité des logiciels, les risques. gestion et génie logiciel. Avant de rejoindre la FDA, il a travaillé 20 ans en tant qu’ingénieur systèmes/logiciels pour des sociétés telles que Ford, Electronic Data Systems, Honeywell et SAIC.
Comment soumettre une contribution à MDO
Les opinions exprimées dans cet article de blog n’engagent que les auteurs et ne reflètent pas nécessairement celles de Medical Design & Outsourcing ou de ses employés.